
Toxic Trouble: Millions of CPAP, BiPAP and ventilators recalled
Philips Respironics is recalling millions of CPAP, BiPAP and ventilators due to potential health risks related to polyester-based polyurethane sound abatement foam.


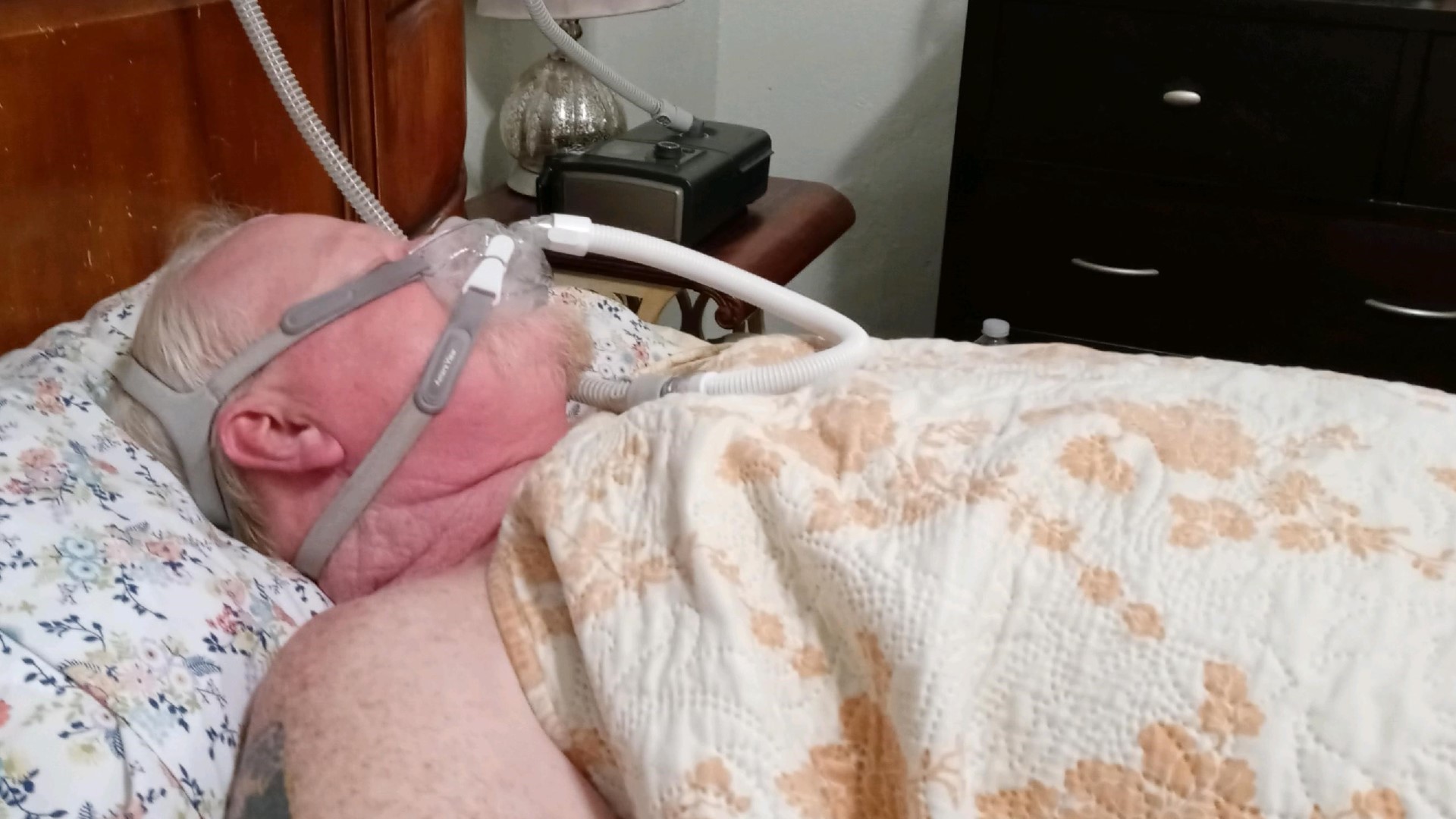
It’s been just over a year since Randy Boyd was diagnosed with cancer. In July 2020, he felt a bruise on the left side of his neck and went to the doctor. A biopsy revealed it was throat cancer.
“I just couldn't believe it,” Boyd said. “And I'm thinking well, how in the heck, can I get cancer out? You know, don't run my family or nothing.”
It's been more than a year of trip after trip to the hospital.
“I ended up doing 33 treatments of radiation and six and a half weeks of chemo, and that took care of the left side,” Boyd said.
Then he had to undergo another surgery because cancer appeared on his right side. He just finished another thirty rounds of radiation.
“I'm struggling, and I'm struggling physically, mentally,” Boyd. “I go to church every Sunday. And I think that's what keeps me from losing it. God keeps me from losing it.”
“It was like a bombshell.”
Boyd was diagnosed with throat cancer at the age of 58, 22 years after he says he stopped smoking.


“I have no idea how I got the cancer, then all sudden, Philips comes out with a recall saying that their machines have a risk of causing cancer, so I'm like, ‘Oh my gosh.' You know, I mean, this could have been the reason that I'm going through all this,’” Boyd said. “It was like a bombshell.”
Like millions of Americans, he was prescribed a Philips CPAP machine to treat sleep apnea. For more than six years, he says he slept with it strapped to his face to help him breathe.
“When I first got a CPAP machine, after the first week, I slept so good. I started dreaming again,” Boyd recalled. “I'd wake up feeling refreshed the next day thinking that you know everything's gonna be great now. And then here six and a half years later, I've got cancer and am having surgeries.”
The Recall
In June, Philips Respironics announced a voluntary recall of millions of its breathing assistance machines, certain ventilators, CPAP and BiPAP machines. They were recalled due to potential health risks related to the polyester-based polyurethane foam used in the devices to make them quieter.
Philips said it received several complaints about the presence of black debris and particles within the airpath circuit and reports of headaches, upper airway irritation, cough, chest pressure and sinus infections.
The urgent recall this summer warned the issues could result in serious injury which can be life-threatening or cause permanent impairment but said there had been no reports of death as a result of these issues.
Attorney Troy Bouk's law firm represents Boyd and thousands of other CPAP users.
“So the problem with the foam in particular is that it does two things. One is it degrades. So, there's small particles that break off and flow through the tube into the mask and ultimately into the person's body,” Bouk said. “And they can either breathe it in into the lungs, or they can digest it and it goes through their digestive system.”
The FDA says the foam issue may get worse in hot and humid settings, and it may also release certain dangerous chemicals.
“When it off-gases, it makes these volatile organic compounds,” Bouk said. “It's those VOCs as we call them, that are harmful to a person that may cause cancer."
The FDA says the potential risks of inhaling or swallowing pieces of foam include toxic or cancer-causing effects to organs such as the kidneys and liver.
In response to the recall, the FDA inspected a Philips Respironics' manufacturing facility. The inspection report just released in November said emails show Philips was made aware of the foam degradation issues in 2015 and did no investigation, health hazard evaluation, risk analysis or design review.
The FDA representative noted in his report a 2018 email from Philips to its supplier documenting complaints Philips received related to problems with the foam in Trilogy ventilator devices. That foam, the report said, had been pulled into patients’ air pathways.
“A follow-up email amongst your firm’s personnel, dated 08/24/2018, states that testing confirmed that the affected foam breaks down in high heat and high humidity environments, which concurred with Trilogy ventilator related complaints received from both Florida and (blurred out). It further states that your firm made the decision not to change the design, and continue to include polyester polyurethane foam, in the Trilogy ventilator platform of devices,” the FDA inspection report said.
It went on to say, “This affected foam was later found to be mutagenic, cytotoxic, carcinogenic, and non-biocompatible.”
According to the FDA, there were at least 14 times where Philips was aware of issues or concerns related to problems with the foam and potentially toxic emissions and each time failed to take corrective action. Between 2014 and 2017, the FDA inspection report says Philips received about 80 complaints related to degraded foam on other CPAP and BiPAP devices.
“We fully understand and regret the impact that this is having on patients. We have launched a comprehensive patient and customer communication program, which includes dedicated mailings, call centers and websites in more than 100 countries,” Mario Fante, Senior Press Officer for Philips said. “This is a complex undertaking and patients may have learned about the recall notification via the news, before they received the direct mailing/letters.”
But some customers may still not be aware of the potential health risks because many of the devices were sold through third parties.
“In the U.S., Philips does not own or manage the CPAP patient’s contact information – many patients purchase their devices through third parties. Philips is working with those third parties (Durable Medical Equipment providers - DMEs) to also reach patients directly as quickly as possible. We are working around the clock to continue to reach out to our customers and patients,” Fante said.
For customers like Boyd who are aware of the potential dangers, getting a replacement device may not happen for months. His is on backorder.

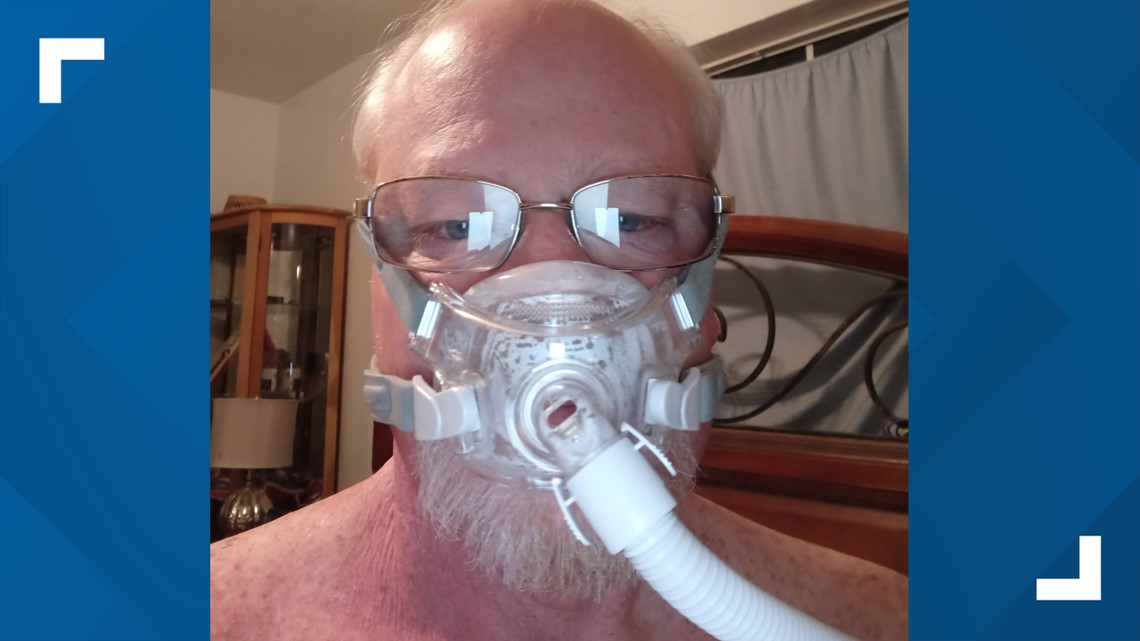
“It's horrible. I mean, I have to put this thing on my face every night and sleep with it, so I make sure I don't die in my sleep. And then when I'm putting it on my face, am I causing the cancer to come back? Is there going to call something to get in my lungs? I have no idea. So, I'm just stuck in the middle. I don't know what to do here,” Boyd said. “I have five kids and twelve grandkids and some of them feel like, ‘Well, Dad, you shouldn't use the machine.’ But then I tell him I have to use the machine because if I don't my doctor says I could stop breathing and die.”
Bouk says he’s hearing from clients around the country who are in a similar situation.
“They’re even calling us how can I get a different machine, but Philips really controlled about 64% of the market in the U.S.” Bouk said. “There's not a lot of large companies that can fill that void of machines that quickly, and so they really don't have a choice. So, they continue to use their machine.”
Philips says its priority is to replace the foam in all the affected devices either by repair or replacement with like devices with new silicone-based foam at no cost to consumers in the United States. That's expected to take until next Fall.
Replacement Foam
According to a FDA news release dated November 12, 2021, “During the manufacturing facility inspection, the FDA obtained additional information, not previously available to the agency, regarding the silicone-based foam used in a singular, similar device marketed outside the U.S., which failed one safety test for the release of certain chemicals of concern, called volatile organic compounds (VOCs). Similar testing provided by Philips Respironics to the FDA on devices authorized for marketing in the U.S. had demonstrated acceptable results.”
The FDA is now requesting Philips Respironics have an independent laboratory perform more tests to determine if the silicone-based foam poses potential safety risks.
For now the FDA is recommending consumers talk to their doctor to decide whether or not they should stop using the recalled device. Philips says you should register your recalled device online to begin the repair/replacement process.
Bouk says the court cases against Philips are being consolidated and heard before a federal judge in Pennsylvania. He expects it will take years to resolve.
“But until we get into discovery and find out what's in the filing cabinets of Philips, we won't really know what's behind the curtain,” Bouk said.
As Philips ramps up its production of replacement devices and repair kits, Boyd waits. He hopes by sharing his story he will spare others from having to go down the difficult road he's traveling.
“Justice for me, they’ve caused me so much pain and so much anguish. My job, with my family, being sick, the pain I've had to go through with the surgeries,” Boyd said. “If this could help somebody else, then it will be worth you know, me speaking up.”
Full statement from Philips to First Coast News:
On June 14, 2021, Philips voluntarily issued a global field safety notice for specific affected devices within its Sleep & Respiratory Care portfolio. The company also issued a global press release on the notice on the same date.
In accordance with medical device regulations and laws in the markets that we serve, Philips is required to broadly communicate a notice of this kind at the earliest possible date. This is intended to ensure wide and timely awareness of potential safety issues as well as important instructions related to clinical use of affected devices.
We fully understand and regret the impact that this is having on patients. We have launched a comprehensive patient and customer communication program, which includes dedicated mailings, call centers and websites in more than 100 countries. This is a complex undertaking and patients may have learned about the recall notification via the news, before they received the direct mailing/letters.
In the U.S., Philips does not own or manage the CPAP patient’s contact information – many patients purchase their devices through third parties. Philips is working with those third parties (Durable Medical Equipment providers - DMEs) to also reach patients directly as quickly as possible. We are working around the clock to continue to reach out to our customers and patients.
To address affected devices, Philips has mobilized the necessary resources across the company to correct the foam component quality issue that we have identified. Our priority is to replace the foam in all the affected devices either by repair or replacement with like devices with the new foam. The repair and replacement program will be at no cost to consumers in the U.S.
Regarding estimated timing, we are working to address this issue as expeditiously as possible. Given the number of devices currently in use (estimated at 3 to 4 million units globally based on production and shipment data – about half are in the U.S.), we expect to complete the repair and replacement programs in each country within approximately 12 months from obtaining the relevant regulatory clearances.
On September 1, 2021, Philips announced the start of the repair and replacement program in the U.S.: Philips starts repair and replacement program - News | Philips.
The replacement of certain affected devices is already underway and Philips anticipates rework of affected devices to commence in the course of this month.
At this time, Philips is already producing repair kits and replacement devices in large quantities. We have increased the production capacity of repair kits and replacement devices in the third quarter of 2021 to 55,000 per week and we aim to further increase that capacity to 80,000 units per week in the fourth quarter of 2021. As our production capacity is fully focused on the repair and replacement actions, we are currently not taking orders for sleep therapy devices for new patients.
We are requesting that patients with affected devices currently in use to register their products on our dedicated web site to facilitate the process. Registration is what formally starts the repair / replacement process.
If a Durable Medical Equipment provider (DME) registers a product for a patient, the DME will manage the replacement process and be in contact regarding the status and expected time frame. If a patient registers their product, Philips and the DME will consult to determine which organization will manage the replacement process. If the DME authorizes Philips to perform the replacement, Philips will be in contact regarding the status and expected time frame. This process is necessary to move forward and may take some time.
Philips continues to recommend that affected patients please follow the advice of their physicians, as they are most familiar with a patient’s medical history. To aid the clinician in the consultation with the patient, Philips has made the following clinical information available: Information for Physicians and other medical care providers | Philips
We will continue to publish updates at the following dedicated web site for the recall: www.philips.com/src-update
Sincere regards,
Mario Fante
Senior Press Officer
Philips Global Press Office